Area M: Modeling and simulation
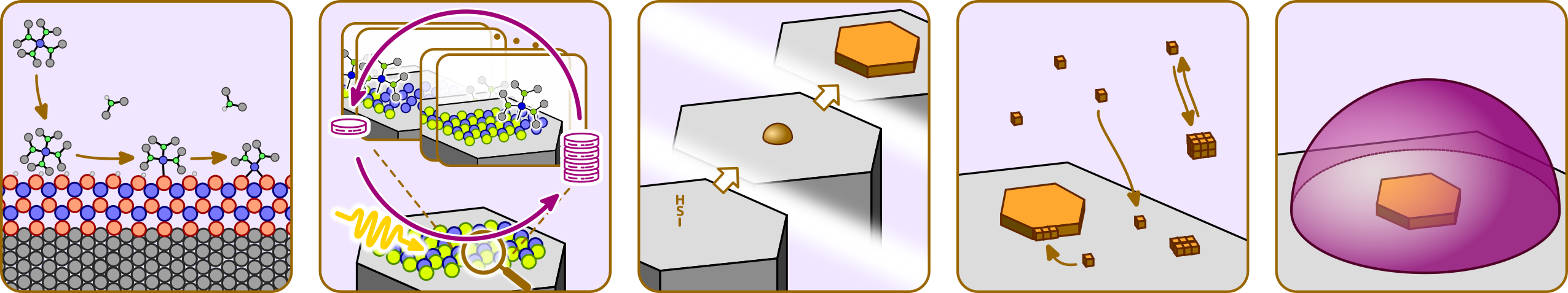
The scientists of Area M set themselves the goal of describing the individual microscopic processes of nucleation and growth as well as macroscopic film formation in an exhaustive manner. The theoretical developments will be based on the close feedback with experimental projects Areas C, S, and F. Describing these inherently heterogeneous events over the relevant time and length scales will be rendered possible by combining a variety of computational approaches:
- the static calculation of interface properties, defect structures, and ground-state electronic properties, the dynamic exploration of reaction coordinates using first-principles density functional approaches, as well as the dynamic description of charge carriers generated upon photoexcitation and subsequent exciton separation (≤10–10 s, ≤10–8 m): M1 (Görling), M2 (Müller);
- the molecular-level modeling of surface reactions and surfactant self-assembly of solutes within explicitly treated solvent with classical molecular dynamics approaches (≤10–7 s, ≤10–6 m): M3 (Zahn);
- the dynamic description, by explicit treatment transport phenomena including the effects of interfacial diffusion on interface reaction mechanisms and deposit morphology (≤10–3 s, ≤10‑2 m): M4 (Smith), and the diffusion within the liquid phase and macroscopic fluid flow at even larger length scales to study the formation of films M5 (Harting).
- Computational data of all Area M projects will be integrated into a materials database. This shared resource will facilitate the development of models for predicting materials with tailored properties.
Project M1 (Görling) applies electronic structure calculations based on density functional theory to obtain insight into dynamics and interaction energies of molecular precursors among themselves and with the surface. The calculation of reaction intermediates and transition states will unravel reaction mechanisms during film formation and surface functionalization (C1, C2, C3, S1).13 Desired reactions will be compared with side-reactions. Surface species and spectroscopic signatures will be identified in collaboration with experimental projects (C4, C5, C6, S1, S4, S5).24,28 Ab initio dynamics will be used to study surface diffusion, calculate vibrational spectra beyond the harmonic approximation, and make direct comparisons with classical MD simulations with parameters provided to M3, M4.
Project leader: Prof. Dr. A. Görling
Project M2 (Müller) develops methods for elucidating excited state phenomena in aggregates of varying sizes, from individual atoms and molecules to 2D semiconductor materials and assembled structures. This project will investigate how alterations in geometry and electronic structure impact spectroscopic data, including electronic absorption and emission spectra. We will address this challenge with a combination of ab initio quantum chemical methods and data science techniques. By integrating computational and experimental results, the project aims to achieve a deeper understanding of photoinduced phenomena, particularly photoswitching reactions (C3) and photoinduced processes in thin films (F1, F4), as examined through time-resolved spectroscopies. The procedures developed will be critically validated through close collaboration with experimental studies (C3, F1, F4, S4).
Project leader: Jun.-Prof. Dr. Carolin Müller
Project M3 (Zahn) provides an atomic-scale description of the dynamics of surface-surfactant-solvent interactions for rationalizing self-assembly steps and reactive events (as in C1, C3, C5). These studies explicitly involve the nascent solid phase, its reorganization upon defects incorporation and the association of molecular reactants and their ligands on the solid surface. They will serve to highlight solvent effects as a tool able to influence the growth morphology in solution-based methods (C1, C5, S3, F3, F6) as opposed to gas-phase techniques. The insight gained by exploiting the Kawska-Zahn molecular dynamics method will guide the design of surface functionalization for ad-layer nucleation and growth, including guides for the stabilization of two-dimensional crystals (C3, F5). Interaction potentials will be benchmarked to M1, M2, while M3 will provide interesting structures as input to M1, M2. M3 will identify fundamental structural features and dynamic processes for M4, M5. It will also rely on a constant comparison with experimental results in S2, S5.
Project leader: Prof. Dr. Dirk Zahn
Project M4 (Smith) provides quantitative kinetic data concerning the non-equilibrium processes of deposition based on statistical methods. A kinetic Monte Carlo framework will allow for appropriate sampling of the statistical ensembles in the open systems used for deposition. The method will be used to model reactions occurring on up to millions of potential surface sites, and determine the chance of nucleation (C1, C2), competitive growth of nucleated seeds, defect appearance, and defect mobility (to be correlated with experimental results from F5). M4 serves as a link within the theory projects, taking input from M1, M2, M3 as to binding energies and electronic structure while providing parametrization input to M5. It also correlates to the chemical information provided by experimental projects C6, S1.
Project leader: Prof. Dr. Ana-Sunčana Smith
Project M5 (Harting) focuses on how physical aspects of the heterogeneous solid/liquid system influence the nucleation and growth occurring at the interface during deposition from dissolved precursors. These aspects include in particular transport (diffusion and flow), concentration inhomogeneities influenced by proximity effects and the potentially three-dimensional nature of the interface and the concentration gradients, as well as solvent evaporation and (de-)wetting. Phase-field methods will be exploited to describe nucleation on patterned substrates (as in C1) and to discover which experimental tools are available to drive two-dimensional growth. Electrostatic energies and specific nucleation sites will also be implemented into the model in order to allow for a direct comparison of M5 predictions with esults of C1, F3, F4, S2, S3, and with support on model parametrization by M3, M4.
Project leader: Prof. Dr. Jens Harting